近年来,细胞与基因疗法(Cell and Gene Therapy, CGT)已逐渐成为最引人注目的领域之一,在带来颠覆性治疗的同时,CGT市场也迎来蓬勃发展。2016-2020年,全球CGT市场规模年复合增长率达 153.3%,2020年为20.75亿美元。CGT 2022年全球市场体量将达 到200亿美元,保持50%的高速增长率。预计到2025年,市场规模有望达到305.39亿美元。虽然中国市场起步较晚,但在整体市场发展的推动下,在政策推动、 医学技术进步、监管日益完善等多重因素影响下,2025年有望激增到 178.85亿元。
图一|全球以及中国CGT的市场规模

在全球及中国生物医药市场均保持高速增长的大背景下,在技术创新不断涌现、供应链优势不断凸显的支撑下,国内生物医药企业开始在海外布局,拓展国际市场。特别是在我国药监部门加入ICH(国际人用药品注册技术协调会)后,为了实现与国际接轨,越来越多的本土药企按照 FDA 的标准研发和申报新药;同时境外的新药也加速在中国落地,这也推动了中美双报的发展。
中美双报是指用同一套研究资料,同时或分阶段在中美两国分别进行申报,同时敲响两地市场的大门。但是“中美双报”并不是简单地把一份资料分别递送给NMPA和FDA,而是在ICH框架下同步开展在两国的新药开发,并在研发中尽可能同时满足两国的法规要求。尽管中美申报法规基本一致,但是仍然有很大的差异。因此要制定相对完善的申报策略,需要企业在申报过程中结合具体的产品特点、患者种族的差异以及中美法规监督部门对申报资料的要求等情况具体分析。本文以中国的II类、美国的Type B沟通交流会议书面回复和新药临床试验申(Investigational New Drug Application ,IND)为例,对两者的区别进行了简单地梳理。
申报流程
相同处:原则上,先与监管方进行pre-IND沟通交流会议,监管方就沟通交流问题做出建议后,申请人根据监管建议,补充相关研究后提交IND申请。
不同处:
中国:
申请人经“申请人之窗”提交《沟通交流会议申请表》*和《沟通交流会议资料》**
CDE在收到申请后3日内完成初步审核,符合要求者送达专业审评团队
II类会议回复时限为60个工作日。经“申请人之窗”推送pre-IND问题答复
IND资料受理后60个工作日,若临床试验申请通过,CDE将经“申请人之窗”推送“临床试验批准通知书”
图二|中国preIND+IND申报流程

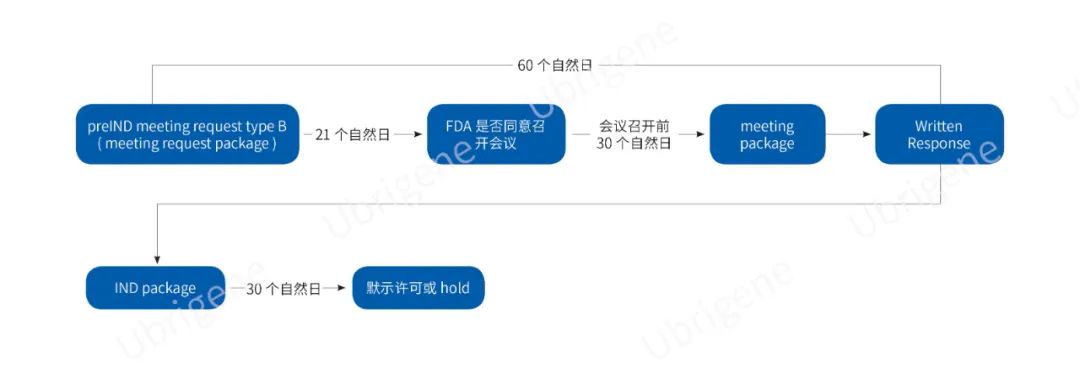
美国:
提前一周申请pre-IND编号
eCTD交preIND Request(问题清单不超过12个问题)后21天收到FDA回信
第30天提供preIND package(所有问题的背景信息,包括所有附件不超过150页)
第60天收到书面回复或电话会议,电话会议将在结束1个月左右收到Meeting Minute
一个产品或多个高度相似的产品只有1次pre-IND的机会,需要进一步沟通的问题可以要求Type D会议,但是Type D只有30%~50%的批准概率
IND提交30天后,FDA不回信就代表通过
图三|FDA pre-IND+IND申报流程
申报资料内容
相同处:格式和内容参考ICH M4 的要求。
不同处:
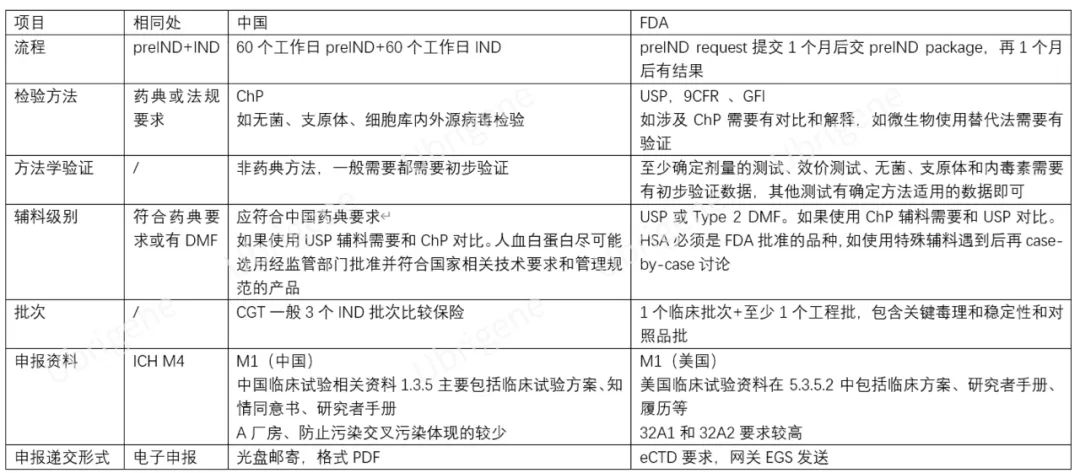
M1是区域行政信息,中美不太一致。
中国的M1包括说明函、申请表、preIND回复、临床药物说明书等。
美国的M1包含行政表格(1571、3674)、US Agent信息、DMF引用信、IB、preIND回复、环境评估(一般IND直接交豁免信)等。
中国临床试验相关资料1.3.5主要包括临床试验方案、知情同意书、研究者手册。
美国临床试验资料则在5.3.5.2体现,包括临床方案、ICF、CV,批准后交1572表格
申报递交形式
中国:以电子形式(光盘)邮寄申报资料,格式为PDF,电子签章。药审中心持续推进对电子申报普及,规范电子光盘技术要求规范。
美国:按照ICH M8要求,采用eCTD电子注册文档的形式。M2、M3S、M3P、M3A需要拆分至合适的粒度、M4毒理报告要求SEND格式、所有报告要有STF。小于等于10G的eCTD可以通过电子提交网关ESG提交。
生产批次
中国:CGT一般3个IND批次,对GMP要求较高。
美国:1个临床批次+至少1个工程批,包含关键毒理和稳定性和对照品批。I期临床批次对GMP要求较低。
生产过程控制
中国:需要中控试验和中间品稳定性方案,可以不提供数据
美国:需要中控和中间品检测的数据,当有中间品储存时要有中间品稳定性数据。
细胞库和病毒库
中国:按照中国药典三部通则《生物制品生产检定用动物细胞基质制备及质量控制》和《生物制品生产检定用菌毒种管理及质量控制》要求执行。
美国:按USP、9 CFR和GFI完成外源因子测试。
辅料级别
中国:应符合中国药典要求。如果使用USP辅料需要和ChP对比。人血白蛋白尽可能选用经监管部门批准并符合国家相关技术要求和管理规范的产品。
美国:应为USP或Type 2 DMF。如果使用ChP辅料需要和USP对比。HSA必须是FDA批准的品种。如果有特殊辅料(比如mRNA用全新结构的脂质体),需要以DMF或在3.2.A.3中递交完整资料。
检验方法
中国:应符合中国药典要求。如无菌、支原体、细胞内外源病毒检验应符合中国药典要求。
美国:应符合美国药典要。如无菌、支原体、细胞内外源病毒检验应符合美国药典、9CFR要求。如涉及ChP需要有对比和解释,如微生物使用替代法需要有验证。
放行标准
相同处:一般包括鉴别、纯度和杂质、含量、生物学活性、安全性和制剂相关指标。
美国:异常毒性(general toxicity)只有在极少的情况下需要。
稳定性
中国:运输稳定性应能覆盖至国内医疗机构的运输方式和时长。
美国:应有运输条件下(运输稳定性应可以模拟运输至国外的运输方式和时长)的稳定性数据,CGT产品需要有给药器械和给药条件下的稳定性数据。
方法学验证
中国:非药典方法,一般需要都需要初步验证。在药品开发初期,不需要提交全面完整的分析方法验证资料,但至少应提供方法的专属性、灵敏度等关键项目的验证信息。
美国:至少确定剂量的测试、效价测试、无菌、支原体和内毒素需要有初步验证数据,其他测试有确定方法适用的数据即可。
包装容器
中国:优先选择在“原辅包登记平台”上登记的包材。
美国:可能的话提供Type III DMF
小结
自2017年我国加入ICH以来,药品注册管理制度加速与国际接轨,也推动了中美双报的发展。中美双报不仅能帮助本土企业的产品在更多国家尤其是发达国家市场上市,为企业提供更大的市场和更多的利益,还可以加快产品研发速度,培养国际化临床研究团队,并大幅度地提升本土企业的国际影响力。
然而,尽管中国NMPA和美国FDA在有着大致相同的审核方向,中美双报仍然有很大的挑战性。
表一|中国的II类、美国的Type B沟通交流会议书面回复和IND申请比较
(来源:医前沿)