1.临床药理学-概况
临床药理学研究贯穿整个药物研发过程,以评价药物安全性和有效性为主要任务。介绍临床药理学前,很有必要介绍下临床分期。
通常,临床研究被分为四个时间周期(I期-IV期);但,分期的概念并不能为临床试验的分类提供足够的根据,因为一类试验可能发生在几个分期中。基于此,分期的概念实际上仅是一种描述,而不是一套要求。
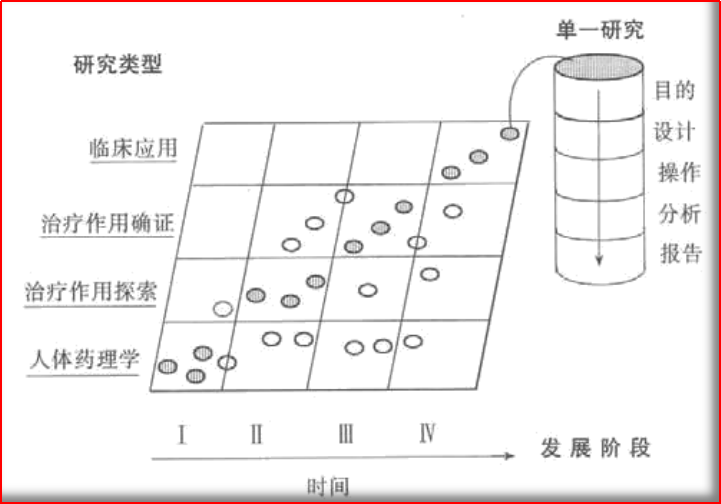
图1.1 研发分期和研究类型间的联系
研究分期的概念并不意味着研究必须按照固定的顺序进行,因为对于某些药物来说,按照常规的顺序进行的研发计划是不适宜或者不必要的。例如:尽管人体药理学研究一般是在I期进行,但在其他III期中也常进行很多此类研究,有时仍会被归入I期研究。
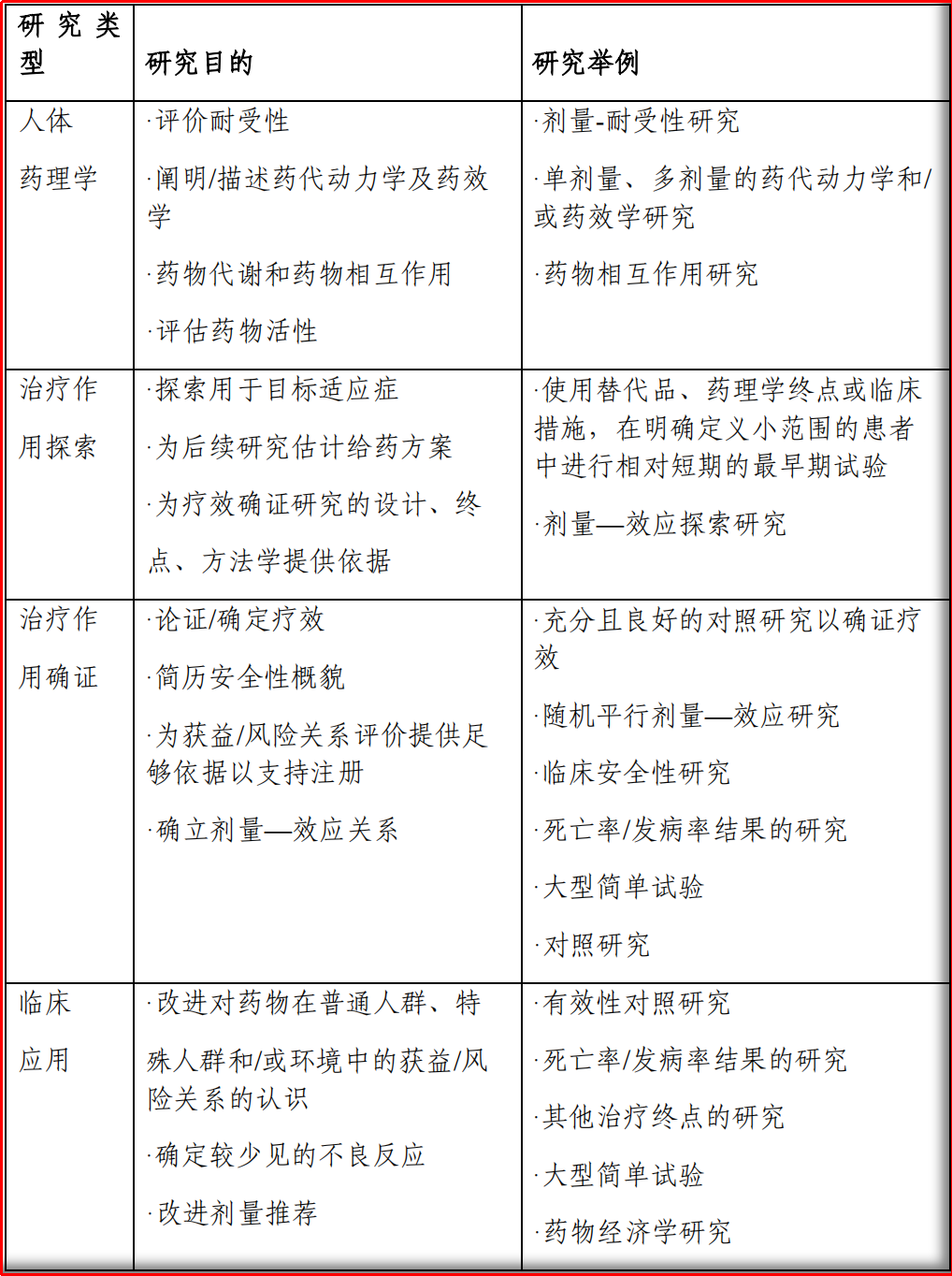
图1.2 根据研究目的对临床研究进行分类
所以,临床药理学更多的是关联于传统的I期临床工作,或者说研发早期的临床试验。值得注意的是,有时在治疗作用探索在早期临床研究阶段就可以进行,此时也可以把临床药理学的研究内容和初步的疗效评估相结合。
基于上述内容,临床药理学的主要工作为:1)在可接受的安全性下,预测人体对在研新药的最大耐受剂量;2)在全剂量范围之内表征剂量限制性的、跟暴露量相关的不良效应;3)如果有合适的生物标志物,还可以在这个范围内表征药代/药效关系;4)争取获得机制验证或概念验证的研究结果。综上,临床药理学研究的核心内容为“剂量-暴露-效应”。
2.以始为终-申报资料对临床药理的要求
创新药上市申报资料中应按照法规要求提交每一项独立临床药理学研究的详细内容,包括研究方案(需提供历次方案的修订版本、修订依据、伦理批件等)、统计分析计划、研究总结报告等。在创新药申报上市申报资料的临床药理学概述性内容中,应提供所有临床药理学研究项目的列表。
首先,应简要介绍已完成的非临床相关研究结果,以帮助阐述药物作用机制(Mechanism of Action,MoA)、解释人体PK、PD(包括疗效和安全性)数据等,比如渗透性、血脑屏障、蛋白结合、肝脏代谢和DDI等。
其次,应简要综述各项人体临床药理学研究结果,比如健康志愿者和/或患者的PK、PD和PK/PD关系的研究,以及内在和外在因素对PK和PK/PD关系的影响等。
再次,应简要阐述研究设计和数据分析的关键信息,比如研究剂量的选择、研究人群、考察的内在或外在因素的选择、PD终点的选择以及采用经典方法或者基于模型等方法收集和分析数据用于评估PK、PD等。
3.临床药理-药代具体工作
根据非临床研究结果,可对创新药在人体内的吸收、分布、代谢等情况进行预测,并可用以解释临床研究结果。比如考察肠吸收和通过血脑屏障能力的渗透性研究、体外蛋白结合研究、肝脏代谢和药物相互作用研究等。
单次/多次给药剂量递增研究
单次给药剂量递增(SAD)研究和多次给药剂量递增(MAD)研究,通常包含安全耐受性评价和PK评价等。其中,单次/多次给药剂量递增PK研究是最早探索创新药人体内PK特征并关联暴露量与药物安全性(有时包含药效)关系的研究,可结合在耐受性研究中开展。
表3.1 SAD/MAD重点工作内容

患者药代动力学研究
患者PK研究,主要研究药物在目标适应症人群中的PK特征,以及患者与健康志愿者(如有)的PK差异。患者PK研究结果为以患者为受试者的探索性和确证性临床研究提供设计依据。患者PK研究有时是独立研究,有时嵌套在评估患者疗效和安全性的探索性和确证性临床研究中。
表3.2 患者药代动力学研究/研究时机

物质平衡研究
物质平衡研究,是考察创新药在人体内的吸收、代谢和排泄特征,以阐明原型药及其代谢产物在人体内代谢/消除的途径和时间过程等问题,其对全面认知创新药的临床用药安全有效性结果具有重要意义。物质平衡研究结果对药物相互作用研究和探索性/确证性临床研究设计具有重要参考作用,对肝/肾功能不全人群研究的必要性提供依据。
PS:建议关注在人体物质平衡研究中发现的未在动物实验中观察到的新代谢产物和与其他物种体内不成比例的高浓度的代谢产物。物质平衡研究可以采用放射性同位素示踪法或其他合适的方法开展。
表3.3 物质平衡研究/研究时机

食物影响研究
食物影响,研究考察的是与不进餐相比,受试者进餐后创新药体内暴露的变化,以及不同类型的饮食对暴露的影响。食物影响研究结果可用以支持后续临床研究中受试者服药和饮食类型或时间的设计安排,并最终用以指导撰写说明书。
PS:食物引起的暴露的变化最终能否对临床用药带来明显影响,需结合临床研究的安全有效性结果以及暴露-效应关系分析进行综合评价。当食物引起的暴露水平的变化对临床用药有明显影响时,需要在说明书中明确患者服药时是否可以同时饮食或者服药和饮食之间的时间窗。如拟上市制剂与临床研究所用制剂不同,建议关注拟上市制剂的食物影响问题。
表3.4 食物影响研究/研究时机

药物相互作用研究
对创新药确定的和可能的产生药物相互作用的因素如代谢酶、转运体等开展临床药物相互作用(DDI)研究,研究结果将指导后续临床研究入排标准、联合用药、剂量调整等设计问题,并将作为说明书中相关内容的撰写依据。
表3.5 药物相互研究/研究时机

肝/肾功能不全患者PK研究
拟开发适应症人群包含肝脏和/或肾脏功能不全患者时应考虑开展相关患者人群的PK研究。建议在创新药临床研发过程中评估肝/肾功能不全对药物PK的影响,以使肝/肾功能不全患者可以考虑被纳入后续临床研究中。肝脏和肾脏作为两大最重要的药物代谢和排泄途径,对多数创新药物尤其是化学药物的PK都可能产生有临床意义的影响。肝功能或肾功能不全的患者对一些创新药体内暴露量可能有一定程度的影响,从而可能影响临床用药安全有效性。
表3.6 肝/肾功能不全患者PK研究/研究时机

生物利用度和生物等效性研究
在创新药临床研究早期阶段,可能通过相对生物利用度研究考察不同的处方、工艺、剂型、规格、给药途径等情况下的暴露量和吸收速率的相似性情况,或结合创新药理化性质和拟开发的目标适应症特点等,不断完善创新药制剂的处方和工艺,为创新药后续开发提供依据。有时需考虑开展绝对生物利用度研究。比如创新药同时开发静脉和非静脉给药剂型时,此时可通过绝对生物利用度研究获得非静脉给药途径的绝对吸收百分数。创新药关键临床研究前甚至上市前(完成关键临床研究后)改变剂型、改变生产场地或放大生产批量等情况时,需按照相关指导原则要求,充分评估其对制剂性能的影响,根据风险评估结果开展研究,必要时需开展生物等效性研究,以支持与此前完成临床研究数据的可桥接性。具体要求建议参考相关技术指导原则。
表3.7 生物利用度和生物等效性研究/研究时机

儿科人群研究
除非拟上市适应症或人群确定不包含儿科患者人群,其他情况通常需在批准儿科人群用药前开展儿科人群研究。研究结果用以指导儿科人群用药方案的制定。儿童的生长发育和疾病状态等因素可能会不同程度影响药物的吸收、分布、代谢和排泄,导致儿童和成人之间、不同年龄段患儿之间的暴露量以及临床的获益和风险均可能不同。因此,在考虑到目标适应症人群年龄的基础上,如必须开展儿科人群研究,应优先在较大年龄段的患儿中开展儿科人群PK研究。儿科人群研究建议参考儿科药物研发的相关法规文件和技术要求。
4.临床药理-药效具体工作
对于临床药理学相关的药效学工作内容,先介绍下表征药物效应的指标,重点包括生物标志物、替代终点、临床终点等。
表4.1 药效指标具体介绍

基于上述药效指标,随着体内研究数据的积累,体外药效学研究数据的权重通常逐渐降低,但是对于某些适应症,体外药效学研究对后期临床研究的意义非常重大。有些特殊情况下无法开展人体研究,体外药效学研究数据可以作为PK/PD分析的支持性证据。PS:药效动力学、QT/QTc间期延长、暴露-效应相关工作如下表所示。
表4.2 药效研究相关的重点项目&内容

5.小结
从2个方面对上述内容进行总结。1)从研究体系理解来说,临床药理学是“剂量-暴露-效应”综合信息的研究过程;SAD/MAD试验即需要开展收集和检测药物有效性和安全性相关指标的工作;药效学指标选取时,应关注所选取指标与临床终点的相关性情况,相关性越强,暴露-效应关系分析结果越可靠,也越有利于后续临床研究尤其是关键临床研究的开展。2)从具体工作融合来说,上述内容为临床药理学的框架式内容,如想真正对其有质的理解,还得需要在具体的项目中进行锻炼,参加每次的项目讨论,对每个参数进行研究,认真撰写申报资料的每句话,并在DSUR中不停的完善研发思路和内容。基于此,慢慢的培养临床药理学在临床试验过程中的具体开展内容和实施时机。
(来源:医前沿)